Abstract
Oropouche virus (OROV) was reported in Cuba in May 2024 and rapidly spread throughout the country. Here, among 147 reverse-transcription polymerase chain reaction-positive cases identified from May to July 2024, we sequenced 39 whole genomes of OROV. Phylogenetic analysis revealed that all sequences formed a monophyletic cluster nested within the reassortant lineage, named OROVBR-2015–2025, which has been circulating extensively in Brazil since 2023. Additional phylogeographic analyses demonstrated that the Cuban subclade probably originated from a single viral introduction from the Brazilian state of Acre in early February 2024, followed by cryptic circulation until its identification in May. The introduction probably occurred in the central region of the country, from which the virus spread and established secondary transmission hubs in the western and eastern regions. These findings underscore the capacity of OROV to spread well beyond South America, which was considered its endemic area of circulation.
Main
Oropouche virus (OROV) was identified in Trinidad and Tobago during the 1950s and later in South and Central America1. OROV generally causes a mild illness, although more severe manifestations such as meningitis, encephalitis, Guillain–Barré syndrome and, more recently, vertical transmission have also been reported2,3. OROV has a single-stranded, negative-sense RNA genome divided into three segments, designated according to their size as L (large, 6.85 kb), M (medium, 4.36 kb) and S (small, 0.95 kb)1. OROV is transmitted between mammals mainly through the bite of infected Culicoides paraensis midges, although Culex quinquefasciatus has been described as a potential secondary vector in urban settings1,4,5.
Since 2024, OROV has spread beyond the Amazon region in Brazil, prompting the Pan American Health Organization (PAHO) to issue an alert regarding its circulation in the Americas6. On 27 May 2024, the Cuban Ministry of Public Health confirmed the detection of local OROV transmission in the country7. Acute febrile Illness (AFI) surveillance in Santiago de Cuba province identified a surge of cases that tested negative for dengue virus. Soon, the Arbovirus National Reference Laboratory at the Institute of Tropical Medicine ‘Pedro Kouri’ identified OROV as the cause of the outbreak. By 28 August, 506 confirmed cases had been reported across 99 out of 168 of the country’s municipalities7.
Recent studies demonstrated that the ongoing OROV outbreak in Brazil resulted from the sustained transmission and dissemination of a OROV reassortant lineage, here named OROVBR-2015–2025, which probably emerged in the Amazonas state between 2010 and 20148 and spread to non-endemic regions of the country across 20249. Preliminary phylogenetic analyses on the S segment of Cuban samples of Santiago de Cuba and Cienfuegos provinces suggested that the Cuban sequences were closely related to 2023–2024 OROV sequences from Brazil10. To better understand the origin and temporality of the first-ever reported OROV introduction in Cuba, as well as the dynamics of its rapid spread throughout the country, we sequenced and analyzed full-length OROV genomes from human serum samples collected between 12 May and 9 July 2024, across 14 of the country’s 16 first-level administrative divisions.
In this period, samples from 217 suspected Oropouche cases were processed by real-time reverse-transcription polymerase chain reaction (RT–PCR)11, and 147 (67.8%) tested positive for OROV mRNA. We selected 39 samples (26.5% of positive cases) for whole-genome sequencing based on Ct values (≤28.5) and geographical and temporal representativeness (Supplementary Table 1 and Extended Data Fig. 1a,b). Maximum likelihood (ML) phylogenetic analyses performed individually for each genomic segment revealed that all OROV sequences belong to the reassortant clade (OROVBR-2015-2025) detected in Brazil8 (Extended Data Fig. 2).
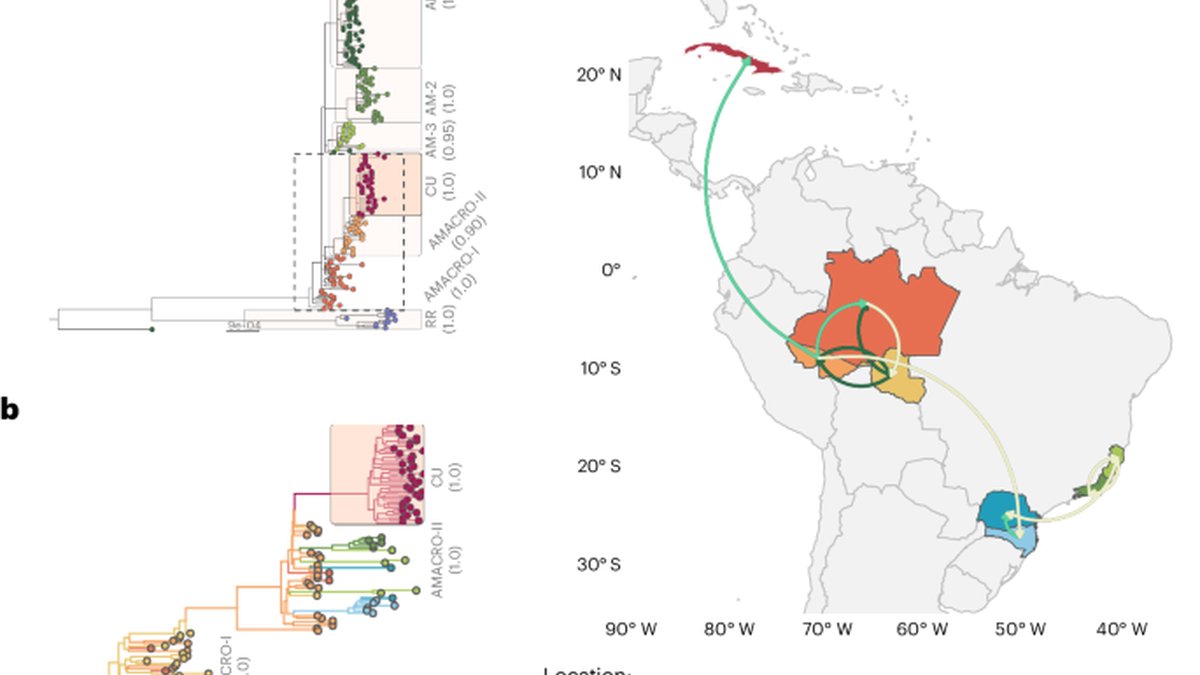
To resolve the phylogenetic relationship between Cuban and Brazilian OROV sequences with more resolution, we conducted a ML phylogenetic analysis on concatenated L, M and S segments of a dataset comprising all OROV sequences from Cuba and sequences representative of Amazonian subclades (AMACROI, AMACROII, AMI, AMII, AMIII and RRI) described previously8. Analysis of the concatenated sequence datasets used in this study revealed no evidence of recombination. This analysis showed that Cuban sequences formed a strongly supported (approximate likelihood-ratio test (aLRT) 1.0) monophyletic cluster (OROV-CU) nested within the Brazilian subclades AMACROI and AMACROII (Fig. 1a). These subclades comprise sequences from Brazilian states distributed across northern (Amazonas, Acre and Roraima), southeastern (Rio de Janeiro and Espírito Santo) and southern (Paraná and Santa Catarina) country regions8. Notably, OROV sequences from cases imported from Cuba in Europe12 also clustered with the OROV-CU subclade (Extended Data Fig. 3 and Supplementary Table 2).
To model the viral diffusion process between Brazil and Cuba, we performed a discrete Bayesian phylogeographic analysis of a dataset of concatenated segments comprising: the oldest sequence of OROVBR-2015–2025 clade detected in the Amazonas state in 2015, all OROV sequences belonging to AMACROI and AMACROII subclades, and the Cuban sequences of this study. This analysis confirms that Cuban sequences branched in a highly supported (posterior probability (PP) 1.0) OROV-CU subclade that most probably originated from a single introduction event from the Brazilian state of Acre (posterior state probability 1.0) (Fig. 1b). The Bayes factor (BF) tests showed significant support for a nonzero rate only between Acre and Cuba (BF 21.3), while connections between other Brazilian locations and Cuba show no significant support (BF <1.5) (Fig. 1c and Supplementary Table 3). The time of the most recent common ancestor (TMRCA) of the OROV-CU subclade was estimated at 2024-02-10 (95% highest posterior density (HPD) 2024-01-04 to 2024-03-17).
We then use geographical coordinates of patient residential areas to reconstruct the fine-scale dispersion of the OROV-CU clade through a continuous spatial diffusion model with non-homogeneous dispersion rates (Fig. 2a). Our analyses indicate that the central provinces, Ciego de Ávila, Sancti Spíritus and Camagüey, served as the principal entry point and early epicenter of spread from early March through early July toward western and eastern regions. The virus dispersed westward via Mayabeque and Artemisa provinces, that seeded transmissions to Pinar del Río and Matanzas provinces in mid-May and to Ciudad de la Habana in late June. The eastern expansion originated through Holguín province, with subsequent dissemination to Santiago de Cuba and Guantánamo provinces in early May. Direct diffusion events from the central region to the province of Pinar del Río were also detected in early July.
Continuous phylogeographic reconstructions revealed that the median distance of viral diffusion events was 22 km (standard deviation 91 km; interquartile range 39 km) (Fig. 2b), remaining relatively constant throughout the study period. Our analysis also demonstrated that very-short-distance events (<2 km) constituted 8% of total viral movements, followed by short-distance (2–10 km, 22%), medium-distance (11–30 km, 33%) and long-distance events (>30 km, 37%). We estimated the OROV-CU median dispersion velocity within Cuba at 1.90 km per day (95% HPD 0.77–3.18 km per day), remaining relatively stable over the study period (Fig. 2c). During the same period interval, coalescent-based population modeling indicates that OROV-CU underwent an initial phase of slow growth until March, a rapid exponential growth from mid-March to mid-April, and a plateau from mid-April to the most recent coalescent event inferred from the tree in late April (Fig. 2d).
These findings confirm that the Cuban OROV outbreak is linked to the spread of the OROVBR-2015–2025 clade, which actively circulates in Brazil, and further reveal that all Cuban sequences were part of a monophyletic cluster (OROV-CU) nested within the Brazilian AMACROII subclade. Our phylogeographic analysis suggests that the Cuban outbreak resulted from a single virus introduction, probably from the Brazilian state of Acre.
The AMACROII subclade circulated in six Brazilian states from Amazonian (Acre and Rondônia) and non-Amazonian (Rio de Janeiro, Espírito Santo, Pernambuco and Santa Catarina) regions. Our findings indicate the virus was introduced to Cuba in early February 2024, concurrent with its arrival in non-Amazonian Brazilian states (mid-January to mid-March)9. This synchronicity makes a direct epidemiological link between these extra-Amazonian states and Cuba unlikely. Conversely, circulation of AMACROII subclade in Acre and Rondônia was traced back to August 20238, providing sufficient time for local spread and subsequent export to Cuba in 2024. Moreover, Acre is a major departure hub for Brazil–Cuba flights (source: Cuban International Health Control Program), providing a plausible route for the AMACROII subclade’s introduction into Cuba. Despite the BF test strongly supporting an epidemiological link between Acre and Cuba, we acknowledge that geographic sampling bias, due to uneven data coverage in the western Amazon and across South America, could confound this result.
Our analysis suggests that the OROV-CU subclade probably arose in early February 2024, indicating a period of approximately 3 months of silent transmission before the virus was detected in Cuba in late May. Our demographic reconstruction supports that the OROV-CU subclade underwent an initial phase of slow expansion until mid-March. Moreover, despite the near-simultaneous detection of OROV transmission in eastern, central and western provinces by the end of May10, our phylogeographic results point to the central provinces as the site of initial introduction. Based on these findings, we propose that the OROV-CU subclade expanded slowly within the central provinces from early February to mid-March, explaining its initial undetected spread. The dissemination of the OROV-CU subclade to the eastern region coincided with a phase of rapid growth, which ultimately led to its detection once a large population size and nationwide spread were achieved.
A previous study of the OROVBR-2015-2025 clade in Brazil estimated an average dispersion rate of 0.66–1.00 km per day across different Amazonian regions and also found that most (65%) dispersal events were over very short distances (<2 km), a pattern consistent with the typical flight range of Culicoides midges8. The OROV-CU subclade seems to have dispersed at a faster average velocity of 1.90 km per day, although the overlapping HPD intervals suggest that the rate estimates in Brazilian Amazon and Cuba are not statistically different. Nevertheless, the proportion of viral movements over distances greater than 10 km was much higher in Cuba (70%) than in the Brazilian Amazon region (30%). This indicates that the mobility of infected individuals was a more important driver of interprovincial viral spread in Cuba.
Our study has some limitations. First, geographic sampling bias can have a large impact on the accuracy of discrete and continuous phylogeographic reconstructions. Although we selected samples to ensure geographical and temporal representativeness, samples with high RT–PCR Ct values were excluded due to the low likelihood of sequencing success and only 26.5% of the cases identified in Cuba during the study period were sequenced. Second, we did not investigate negative dengue samples collected before the recognition of OROV in Cuba. The potential misdiagnosis of early OROV cases therefore represents a possible source of error in the temporal reconstruction. Finally, the primary vector of OROV in Cuba remains unconfirmed. While we have detected the virus in pools of Culex quinquefasciatus mosquitoes and of Ceratopogonidae spp. midges10, and the known vector Culicoides paraensis is present in the country13, the precise role of each species in the outbreak remains uncertain.
The autochthonous transmission of OROV in Cuba demonstrates the virus’s capacity to spread beyond South American areas traditionally considered endemic. This raises concerns about the potential involvement of unrecognized vectors. Given that environmental, climatic and demographic changes are global phenomena, it is not surprising that OROV could spread further across the Americas and beyond, being crucial to strengthen global efforts in enhancing surveillance, improving vector control strategies and advancing research into the factors driving virus transmission.
Methods
Ethics and inclusion
This study was designed through a partnership among the Institute of Tropical Medicine, Pedro Kouri (Cuba), the Instituto Leônidas e Maria Deane, Fiocruz, (Manaus, Brazil) and the Instituto Oswaldo Cruz, Fiocruz (Rio de Janeiro, Brazil), with the coordination and support of the Cuban Ministry of Health and the PAHO. Roles and responsibilities were agreed upon among collaborators ahead of the research, including having both a Cuban and a Brazilian researcher as a co-principal investigators.
The samples processed were obtained anonymously from excess material remaining after routine arbovirus testing, according to the case definition established by the Cuban Ministry of Health as part of the national clinical–epidemiological and laboratory surveillance to identify OROV transmission. Sex/gender information was not considered. Patient personal information was not used or published. Ethics approval was obtained from the Institute of Tropical Medicine, Pedro Kouri (CEI-IPK 54-24).
Previous work from this region (both from our team) was used to guide the design of this study, as well as connect our findings to similar research, and has been taken into account in the citations for this Brief Communication.
Case definition and studied samples
A clinically suspected Oropouche fever case is defined as a patient who resides in or comes from an endemic area or an area with active OROV transmission, or an area with an unusual increase of AFI cases that report fever or fever history (38 °C or more) and headache, accompanied by one or more symptoms as myalgia, arthralgia, chills, low back pain, photophobia or conjunctivitis, with illness resolution in 2–4 days in a favorable manner, with no focalization; or as a patient classified as AFI up to day 6 after symptom onset with a negative dengue IgM test. Suspected cases with a positive real-time RT–PCR result for OROV are considered as confirmed Oropouche cases. Between 12 May and 9 July 2024, samples from 217 suspected Oropouche cases were sent to the Arbovirus-NRL for molecular testing. Real-time RT–PCR confirmed 147 Oropouche cases; 39 (26.5%) were selected for genomic characterization based on cycle threshold (Ct) values (≤28.5), geographical representativeness and chronological distribution of sample collection by epidemiological week. Anonymized patient information data were used and are provided in Supplementary Table 1. Samples were collected from patients from 14 out of 15 provinces except for Las Tunas province and Isla de la Juventud municipality (both with no positive samples) (Cuban territory comprises 15 provinces (main island) plus the special municipality of Isla de la Juventud (a small island near the main island)). Among the 39 infected individuals, 33% (13/39) were male and 67% (26/39) were female, with a median age of 49 years (range 12–90 years). None of the 147 cases detected during the study period presented severe clinical manifestations.
RT–PCR testing for dengue, Mayaro and Oropouche viruses
Country-wide laboratory diagnosis of OROV infection was carried out by the Arbovirus-NRL. Acute serum samples collected 1 to 7 days after symptom onset were extracted using the QIAamp Viral RNA Mini Kit (Qiagen), according to the manufacturer’s instructions. RNA extracts were first tested for dengue by RT–PCR14. Dengue-virus-negative samples were tested using a multiplex real-time protocol designed to amplify OROV and Mayaro virus RNA.
OROV whole-genome sequencing and genome assembling
OROV genome amplification and sequencing libraries were prepared with COVIDSeq kit (Illumina) using OROV-specific primers on 2× 150 cycles paired-end runs on a MiSeq instrument (Illumina), as previously described8. Raw sequencing data were converted to FASTQ files on Illumina BaseSpace (https://basespace.illumina.com FASTQ files were imported to Geneious Prime v2024.0.7, trimmed and assembled using a customized workflow. The GenBank sequences OL689332, OL689333 and OL689334 were templates for the S, M and L OROV segments, respectively.
OROV whole-genome genotyping
Complete OROV consensus sequences of L, M and S segments of all 39 newly generated genomes from Cuba were aligned with corresponding segments of a dataset of 92 sequences containing the prototype sequences of OROV (L: AF484424; M: AF441119; S: AY237111), Iquitos virus (L: KF697142; M: KF697143; S: KF697144), Perdões virus (KP691627; M: KP691628; S: KP691629) and Madre de Dios virus (L: KF697147; M: KF697145; S: KF697146), as well as OROV sequences representing the major M (M1 and M2), L (L1 and L2) and S (S1, S2 and S3) clades8 (Supplementary Table 2). Sequences were aligned using MAFFT v715 under the automatic selection of the appropriate strategy according to data size and submitted to phylogenetic reconstruction by ML using IQ-TREE v2.1.116, under the GTR + G4 + I model as selected by the ModelFinder17 software. The tree branches’ support was evaluated with Shimodaira–Hasegawa aLRT (SH-aLRT)18, with 1,000 replicates. ML phylogenetic trees were visualized and edited with FigTree v1.4.4 (https://github.com/rambaut/figtree/releases
Recombination analysis
Alignments of concatenated S, M and L genomic segments were constructed for different OROVBR-2015–2025 datasets and screened for potential recombination using the Recombination Detection Program (RDP) version 519. The recombination breakpoints and potential parental sequences of recombinants were analyzed using nine different detection algorithms (RDP, Geneconv, Bootscan, Maxchi, Chimaera, SiSscan, PhylPro, LARD and 3Seq) implemented in the RDP5 package, with default settings. Only recombination events detected by at least three different methods and P values less than 0.05 were considered reliable.
OROV lineage identification
To identify the OROVBR-2015-2025 lineage(s) circulating in Cuba, we generated a concatenated alignment of genomic segments L, M, and S, comprising all 39 newly generated genomes from Cuba along with a subset (n = 224) of sequences representative of the Amazonian subclades (AMAROI, AMACROII, AMI, AMII, AMIII and RRI) previously described8 (Supplementary Table 2). The resulting dataset (n = 263) was aligned and submitted to ML phylogenetic reconstruction as described above, under the GTR + G4 + I model as selected by the ModelFinder17 software. The tree branches’ support was evaluated with SH-aLRT18, with 1,000 replicates, and the ML phylogenetic tree was visualized and edited with FigTree v1.4.4 (https://github.com/rambaut/figtree/releases
Discrete Bayesian phylogeographic analyses
To infer the spatiotemporal dynamics of the OROV Cuban epidemic, we generated a concatenated alignment of genomic segments L, M and S comprising: (1) all 39 newly generated genomes from Cuba; (2) all available sequences of the closely related Brazilian subclades AMACROI (n = 34) and AMACROII (n = 43) in which Cuban sequences were nested; and (3) the earliest OROVBR-2015–2025 sequence sampled in the Amazonas state in 2015 (Supplementary Table 2). The resulting dataset (n = 117) was aligned and submitted to ML phylogenetic reconstruction as described above. The temporal structure of the ML tree was estimated by performing a root-to-tip linear regression with TempEst v1.5.320, and the significance of the correlation between the collection date and genetic distance was evaluated using a Pearson correlation test, with corrections applied for multiple comparisons. The time-scaled phylogeographic tree was reconstructed using the Bayesian Markov chain Monte Carlo (MCMC) approach, implemented in the software BEAST 1.1021 with BEAGLE library v422, to improve computational time. Bayesian trees were reconstructed using the GTR + G4 + I nucleotide substitution model, the nonparametric Bayesian Skyline coalescent demographic model23 and a relaxed molecular clock model with a continuous-time Markov chain rate reference prior24. The spatial diffusion from Brazil to Cuba was modeled using an asymmetric discrete phylogeographic model with a continuous-time Markov chain rate reference prior24. The most relevant migration pathways between locations were identified by applying the Bayesian stochastic search variable selection approach25, and significant nonzero rates were summarized using the cross-platform SPREAD application26. We consider rates yielding a BF >3 as well-supported diffusion rates. In this analysis, Cuban sequences were assigned to the national level, while Brazilian sequences were classified at the subnational state level. MCMC simulations were run sufficiently long to ensure convergence (effective sample size >200) in all parameters as assessed in TRACER v1.727. The maximum clade credibility (MCC) trees were summarized with TreeAnnotator v1.10 and visualized using ggtree v3.2.1 R packages28.
Continuous Bayesian phylogeographic analyses
To investigate the OROV dissemination dynamics within Cuba, a continuous phylogeographic analysis was conducted with a concatenated dataset consisting exclusively of the Cuban sequences. The viral diffusion process was reconstructed using software BEAST 1.10 as described above, with a relaxed random walk phylogeographic model with a Cauchy distribution29, and an informative prior on root height inferred from the previous discrete phylogeographic analysis (normal distribution, mean 0.30, standard deviation 0.001). We assigned latitude and longitude coordinates to each sequence based on the patient’s place of residence (municipality), adding a slight jitter (0.01 to both coordinates) to tips sharing identical coordinates. Convergence of MCMC chains was accessed as described above. The SPREAD v1.0.7 software30 was used to summarize migratory events and reconstruct the spatiotemporal diffusion patterns of the virus. These patterns were then visualized on maps generated using the ggplot231 and naturalearth32 R packages. In addition, the branch dispersal velocity of the Cuban cluster was inferred from a subsample of 1,000 trees using the seraphim R package33.
Statistical analyses
No statistical method was used to predetermine sample size. No data were excluded from the analyses. Statistical analyses of spatiotemporal and dissemination dynamics were conducted as described in the ‘Discrete Bayesian phylogeographic analyses’ and ‘Continuous Bayesian phylogeographic analyses’ sections.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All the OROV genomes generated in this study were deposited at GISAID under accession numbers EPI_ISL_19611792–EPI_ISL_19611830 (https://doi.org/10.55876/gis8.260330dk (ref. 34). Sample data can be found in Supplementary Table 1, and the sequence data used for phylogenetic analyses are provided in Supplementary Table 2. For further information, please contact the corresponding authors.
References
Sakkas, H., Bozidis, P., Franks, A. & Papadopoulou, C. Oropouche fever: a review. Viruses 10, 175 (2018).
de Armas Fernández, J. R. et al. Report of an unusual association of Oropouche fever with Guillain–Barré syndrome in Cuba, 2024. Eur. J. Clin. Microbiol. Infect. Dis. 43, 2233–2237 (2024).
Oropouche: Cases of Mother-to-Child Transmission under Investigation in Brazil (PAHO, 2024).
Travassos da Rosa, J. F. et al. Oropouche virus: clinical, epidemiological, and molecular aspects of a neglected orthobunyavirus. Am. J. Trop. Med. Hyg. 96, 1019–1030 (2017).
Feitoza, L. H. M. et al. Influence of meteorological and seasonal parameters on the activity of Culicoides paraensis (Diptera: Ceratopogonidae), an annoying anthropophilic biting midge and putative vector of Oropouche virus in Rondônia, Brazilian Amazon. Acta Trop. 243, 106928 (2023).
Epidemiological Alert - Oropouche in the Americas Region - 13 December 2024 (PAHO, 2025).
Nota informativa del Ministerio de Salud Pública 27 de mayo de 2024 (MINSAP, 2024).
Naveca, F. G. et al. Human outbreaks of a novel reassortant Oropouche virus in the Brazilian Amazon region. Nat. Med. 30, 3509–3521 (2024).
Gräf, T. et al. Expansion of Oropouche virus in non-endemic Brazilian regions: analysis of genomic characterisation and ecological drivers. Lancet Infect. Dis. 25, 379–389 (2024).
Benitez, A. J. et al. Oropouche fever, Cuba, May 2024. Emerg. Infect. Dis. 30, 2155–2159 (2024).
Naveca, F. G. et al. Multiplexed reverse transcription real-time polymerase chain reaction for simultaneous detection of Mayaro, Oropouche, and Oropouche-like viruses. Mem. Inst. Oswaldo Cruz 112, 510–513 (2017).
Deiana, M. et al. Full genome characterization of the first Oropouche virus isolate imported in Europe from Cuba. Viruses 16, 1586 (2024).
Pérez, Y. M. et al. First report of Culicoides paraensis (Goeldi, 1905) (Diptera: Ceratoponidae) in Cuba: a new challenge for public health. Parasite Epidemiol. Control 29, e00423 (2025).
Santiago, G. A. et al. Analytical and clinical performance of the CDC real time RT–PCR assay for detection and typing of dengue virus. PLoS Negl. Trop. Dis. 7, e2311 (2013).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Shimodaira, H. & Hasegawa, M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 16, 1114–1116 (1999).
Martin, D. P. et al. RDP5: a computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 7, veaa087 (2021).
Rambaut, A., Lam, T. T., Max Carvalho, L. & Pybus, O. G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2, vew007 (2016).
Suchard, M. A. et al. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4, vey016 (2018).
Suchard, M. A. & Rambaut, A. Many-core algorithms for statistical phylogenetics. Bioinformatics 25, 1370–1376 (2009).
Drummond, A. J., Rambaut, A., Shapiro, B. & Pybus, O. G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22, 1185–1192 (2005).
Ferreira, M. A. R. & Suchard, M. A. Bayesian analysis of elapsed times in continuous-time Markov chains. Can. J. Stat. 36, 355–368 (2008).
Lemey, P., Rambaut, A., Drummond, A. J. & Suchard, M. A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 5, e1000520 (2009).
Bielejec, F. et al. SpreaD3: interactive visualization of spatiotemporal history and trait evolutionary processes. Mol. Biol. Evol. 33, 2167–2169 (2016).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G. & Suchard, M. A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904 (2018).
Yu, G., Smith, D., Zhu, H., Guan, Y. & Lam, T. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36 (2016).
Lemey, P., Rambaut, A., Welch, J. J. & Suchard, M. A. Phylogeography takes a relaxed random walk in continuous space and time. Mol. Biol. Evol. 27, 1877–1885 (2010).
Bielejec, F., Rambaut, A., Suchard, M. A. & Lemey, P. SPREAD: spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics 27, 2910–2912 (2011).
Wickham, H. ggplot2. Elegant Graphics for Data Analysis (Springer, 2016).
rnaturalearth. GitHub https://github.com/ropensci/rnaturalearth (2024).
Dellicour, S., Rose, R. & Pybus, O. G. Explaining the geographic spread of emerging epidemics: a framework for comparing viral phylogenies and environmental landscape data. BMC Bioinformatics 17, 82 (2016).
Cuban OROV sequences. GISAID https://doi.org/10.55876/gis8.260330dk (2024).
Acknowledgements
This study is supported by the Cuban Ministry of Health¸ FAPEAM Call 04/2022/FIOCRUZ/FAPEAM/FAPERO – INOVAÇÃO NA AMAZÔNIA, F.G.N.; FAPEAM Call 023/2022 – INICIATIVA AMAZÔNIA + 10, F.G.N.; Conselho Nacional de Desenvolvimento Científico e Tecnológico – CNPq Institutos Nacionais de Ciência e Tecnologia (INCT – VER) e Chamada CNPq/MCTI 10/2023 – Faixa B – Grupos Consolidados – Universal 2023 (421620/2023-4); and the PAHO. The funders of the study had no role in the study design, data collection, data analysis, data interpretation or writing of the report.
Author information
Authors and Affiliations
Contributions
M.G.G. and F.G.N. contributed to study conception and study design. R.G., M.M.P., M.A., L.P., A.J.B., D.H., S.S., V.S., M.M., G.B., I.A., F.G.N. and M.G.G. contributed to laboratory work. R.G., M.M.P., M.A., L.P., A.J.B., D.H., S.S., J.R.d.A., C.P., M.R., L.F., L.G., V.S., M.M., G.B., I.A., S.R., V.K., F.G.N. and M.G.G. contributed to data analysis. R.G., V.S., G.B., I.A. and F.G.N. contributed to results visualization. G.B., I.A., F.G.N. and M.G.G. contributed to writing the original manuscript. R.G., M.M.P., M.A., L.P., A.J.B., D.H., S.S., J.R.d.A., C.P., M.R., L.F., J.L., L.G., V.S., M.M., G.B., I.A., S.R., V.K., F.G.N. and M.G.G. contributed to manuscript revision. M.G.G., F.G.N., L.F., L.G. and J.M. contributed to resources.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Xiang Ji and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Lia Parkin, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Oropouche cases in Cuba and OROV genomes by epidemiological week and province.
a) The blue bars represent the number of Oropouche cases throughout the country, whereas the green bars represent the number of genomes generated in this study for each Epidemiological Week (EW). Therefore, OROV genomes were represented during all EW of the OROV Cuban outbreak. b) The blue bars represent the number of Oropouche cases by province until 9 July 2024, whereas the green bars represent the number of genomes generated in this study for each province.
Extended Data Fig. 2 ML phylogenetic analyses of the M, L, and S segments of Cuban OROV sequences.
Phylogenetic trees were constructed based on the M (a), L (b), and S (c) segments of 131 complete OROV genomes, including the 39 genomes generated in this study and 92 prototypical sequences representing other OROV lineages from different countries and years, as suggested by Naveca et al.8. The OROVBR-2015-2025 clade is highlighted in light gray in each tree, and its statistical support (SH-aLRT) is annotated. Tree branches are drawn to genetic distance, as indicated by the scale bar at the bottom of each panel. Cuban and prototypical OROV sequences are represented in colors following the coding scheme shown in the left panel and their genotypic classification.
Extended Data Fig. 3 International Dissemination of OROV-CU.
Maximum likelihood phylogenetic tree based on concatenated OROV genomes (n = 265) from the OROVBR-2015-2024 clade, including Cuban OROV genomes (n = 39), and two OROV genomes sampled in Italy (n = 1) and the Netherlands (n = 1). Clusters are annotated with their respective names and statistical support values (aLRT). The tree is scaled according to the legend at the bottom of the panel.
Supplementary information
Supplementary Information (download PDF )
Supplementary Tables 1 and 3.
Supplementary Table 2 (download XLSX )
Metadata for complete OROV genomes from the Brazilian Amazon Region, Cuba and Europe (L, M and S segments).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article
Cite this article
Gravier, R., Perez, M.M., Álvarez, M. et al. Spatiotemporal diffusion of the 2024 Oropouche outbreak in Cuba. Nat Med (2026). https://doi.org/10.1038/s41591-026-04411-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41591-026-04411-9